
购物车0
产品总数:61763


| 中文名称 |
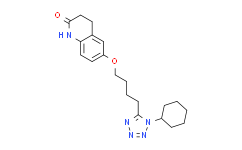
Cilostazol.
|
||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 中文别名 |
西洛他唑;6-[4-(1-环己基-1H-戊四唑-5-基)丁氧基]-3,4-二氢-2(1H)-喹诺酮;西斯他唑;四氮唑(西洛他唑中间体);6-[4-(1-环己基-1H-四唑-5-基)丁氧基]-3,4-二氢-2(1H)-喹诺酮;2,3-二氯吡嗪;6-[4-(1-环己基-1H-四氮唑-5-基)丁基]3,4-二氢喹啉-2-(1H)-酮;Cilostazol 西洛他唑;西洛他唑 Cilostazol;西洛他唑 USP标准品;西洛他唑标准品(JP);6-[4-(1-环己基-1H-四唑-5-基)丁氧基]-3,4-二氢-2(1H)-喹啉酮;雷公藤红素;雷公藤红素,南蛇藤素,苦瓜苷
|
||||||||||||
| 英文名称 |
Cilostazol
|
||||||||||||
| 英文别名 |
Cilostazol;CILOSTAZOL INTERMEDIATES;6-[4-(1-CYCLOHEXYL-1H-TETRAZOL-5-YL)BUTOXY]-3,4-DIHYDRO-2(1H)-QUINOLINONE;6-[4-(1-cyclohexyl-1h-tetrazol-5-yl)butoxy]-3,4-dihydro-2(1h)-quinolinone additional name: 6-[4-(1-cyclohexyl-5-tetrazolyl)butoxy]-1,2,3,4- tetrahydro-2-oxoquinolinone;CILASTAZOL;CILOSTAZOLE;RETAL;OPC 13013;OPC 21;6-(4-(1-Cyclohexyl-1H-tetrazol-5-yl)butoxy)-3,4-dihydroquinolin-2(1H)-one;Cilostal;CILOSTAZOL JP;PLETAAL;PLETAL;6-[4-(1-Cyclohexyl-1H-tetrazol-5-yl)-butoxy]-3,4-dihydro-2(1H)-quinolinone
|
||||||||||||
| Cas No. |
73963-72-1
|
||||||||||||
| 分子式 |
C20H27N5O2
|
||||||||||||
| 分子量 |
369.46
|
||||||||||||
| 包装储存 |
|
||||||||||||
| 详情描述 |
PDE3抑制剂,Cilostazol(OPC 13013; OPC 21)是存在于心血管系统中的PDE3A抑制剂,IC50为200 nM。 |
| 生物活性 |
Cilostazol (OPC 13013) is a potent and selective inhibitor of phosphodiesterase (PDE) 3A, the isoform of PDE 3 in the cardiovascular system, with an IC50 of 0.2 μM. |
||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 性状 |
Solid |
||||||||||||||||
| IC50 & Target[1][2] |
IC50: 0.2 μM ( PDE 3A) |
||||||||||||||||
| 体外研究(In Vitro) |
Cilostazol selectively inhibits cGMP-inhibited phosphodiesterase (PDE 3) and is a potent inhibitor of platelet aggregation induced by various agonists. Medlife has not independently confirmed the accuracy of these methods. They are for reference only. |
||||||||||||||||
| 体内研究(In Vivo) |
Cilostazol (clinically used doses; p.o.; for 2 weeks) could alleviate CCl4 -induced hepatic fibrogenesis in vivo, presumably due to its direct effect to suppress HSC activation. Medlife has not independently confirmed the accuracy of these methods. They are for reference only.
|
||||||||||||||||
| 运输条件 |
Room temperature or refrigerated transportation. |
||||||||||||||||
| 储存方式 |
|
||||||||||||||||
| ClinicalTrial |
|
||||||||||||||||
| 参考文献 |
|
| 溶解度数据 |
体外研究:
DMSO : 50 mg/mL (135.33 mM; Need ultrasonic) 配制储备溶液
*
产品不同,其溶解度不同。建议根据产品选择合适的溶剂配制储备溶液;配成溶液后,建议分装保存,避免反复冻融造成的产品失效。 体内研究:
建议根据您的实验动物和给药方式选择适当的溶解方案。以下溶解方案都建议先按照 体外研究 方式配制澄清的储备液,再依次添加助溶剂:
——为保证实验结果的可靠性,澄清的储备液可以根据储存条件,适当保存;体内实验的工作液,建议您现用现配,当天使用;
以下溶剂前显示的百
*
|
|---|
[1]. Schr?r K. The pharmacology of cilostazol. Diabetes Obes Metab. 2002 Mar;4 Suppl 2:S14-9. [Information]
[2]. Minami N, et al. Inhibition of shear stress-induced platelet aggregation by cilostazol, a specific inhibitor of cGMP-inhibited phosphodiesterase, 体外研究 and ex vivo. Life Sci. 1997;61(25):PL 383-9. [Information]
[3]. Saito S, et al. Cilostazol attenuates hepatic stellate cell activation and protects mice against carbon tetrachloride-induced liver fibrosis. Hepatol Res. 2013 Apr 19. [Information]
[4]. Ye YL,et al. Cilostazol, a phosphodiesterase 3 inhibitor, protects mice against acute and late ischemic brain injuries.Eur J Pharmacol. 2007 Feb 14;557(1):23-31. Epub 2006 Nov 10. [Information]
1:一般建议:溶解度为Medlife测试数据,可能与文献描述存在差异。这是由于生产工艺和批次不同产生的正常现象。为了使其更好的溶解,请用37℃加热试管并在超声波水浴中震动片刻。不同批次产品溶解度各有差异,仅做参考,具体以实验方案为准。
2:储存条件:粉末-20°C一般情况可以保存3年,溶于溶剂-80°C一般情况可以保存1年。不同产品及不同批次产品可能存在差异,请细致阅读产品信息,并辅助参考相关文献描述。
Copyright © 2025 陌孚医药 All rights reserved 未经授权禁止拷贝本站所有资料,如有违反,将追究法律责任。
 沪ICP备2023012080号 |
沪ICP备2023012080号 | ![]() 沪公网安备31011402010657号
沪公网安备31011402010657号
 扫码关注公众号
扫码关注公众号
 产品使用指南
产品使用指南
